电子商务网站建设需求说明书企业定制
之前看过一篇文章,说一个工作多年的老员工,处理数据时只会用复制粘贴到 Excel ,天天加班工作还完不成,后来公司就招了一个会 Python 的新人,结果分分钟就处理完成。所以工作中大家经常会使用 Excel 去处理以及展示数据,但是对于部分工作我们可以借助程序帮忙实现,达到高效解决问题的效果,今天就为大家介绍一下,使用 Python 如何操作 Excel 。
常用工具
数据处理是 Python 的一大应用场景,而 Excel 又是当前最流行的数据处理软件。因此用 Python 进行数据处理时,很容易会和 Excel 打起交道。得益于前人的辛勤劳作,Python 处理 Excel 已有很多现成的轮子,比如 xlrd & xlwt & xlutils 、 XlsxWriter 、 OpenPyXL ,而在 Windows 平台上可以直接调用 Microsoft Excel 的开放接口,这些都是比较常用的工具,还有其他一些优秀的工具这里就不一一介绍,接下来我们通过一个表格展示各工具之间的特点:
| 类型 | xlrd&xlwt&xlutils | XlsxWriter | OpenPyXL | Excel开放接口 |
|---|---|---|---|---|
| 读取 | 支持 | 不支持 | 支持 | 支持 |
| 写入 | 支持 | 支持 | 支持 | 支持 |
| 修改 | 支持 | 不支持 | 支持 | 支持 |
| xls | 支持 | 不支持 | 不支持 | 支持 |
| xlsx | 高版本支持 | 支持 | 支持 | 支持 |
| 大文件 | 不支持 | 支持 | 支持 | 不支持 |
| 效率 | 快 | 快 | 快 | 超慢 |
| 功能 | 较弱 | 强大 | 一般 | 超强大 |
以上可以根据需求不同,选择合适的工具,现在为大家主要介绍下最常用的 xlrd & xlwt & xlutils 系列工具的使用。
xlrd & xlwt & xlutils 介绍
xlrd&xlwt&xlutils 顾明思意是由以下三个库组成:
-
xlrd:用于读取 Excel 文件;
-
xlwt:用于写入 Excel 文件;
-
xlutils:用于操作 Excel 文件的实用工具,比如复制、分割、筛选等;
安装库
安装比较简单,直接用 pip 工具安装三个库即可,安装命令如下:
$ pip3 install xlrd xlwt xlutils安装完成提示 Successfully installed xlrd-1.2.0 xlutils-2.0.0 xlwt-1.3.0 即表示安装成功。
写入 Excel
接下来我们就从写入 Excel 开始,话不多说直接看代码如下:
# excel_w.py# 导入 xlwt 库import xlwt# 创建 xls 文件对象wb = xlwt.Workbook()# 新增两个表单页sh1 = wb.add_sheet('成绩')sh2 = wb.add_sheet('汇总')# 然后按照位置来添加数据,第一个参数是行,第二个参数是列# 写入第一个sheetsh1.write(0, 0, '姓名')sh1.write(0, 1, '成绩')sh1.write(1, 0, '张三')sh1.write(1, 1, 88)sh1.write(2, 0, '李四')sh1.write(2, 1, 99.5)# 写入第二个sheetsh2.write(0, 0, '总分')sh2.write(1, 0, 187.5)# 最后保存文件即可wb.save('test_w.xls')
然后执行命令 python excel_w.py 运行代码,结果会看到生成名为 test_w.xls 的 Excel 文件,打开文件查看如下图所示
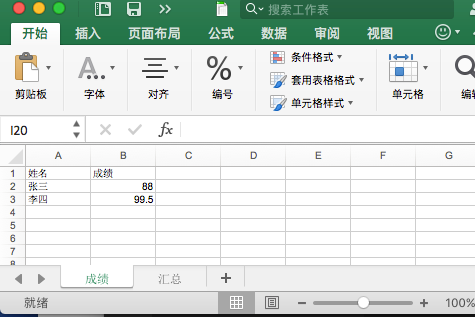
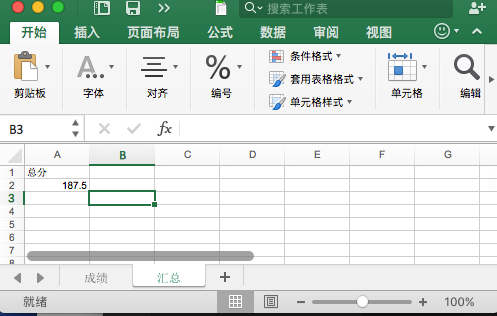
以上就是写入 Excel 的代码,是不是很简单,下面我们再来看下读取 Excel 该如何操作。
读取 Excel
读取 Excel 其实也不难,请看如下代码:
# excel_r.py# 导入 xlrd 库import xlrd# 打开刚才我们写入的 test_w.xls 文件wb = xlrd.open_workbook("test_w.xls")# 获取并打印 sheet 数量print( "sheet 数量:", wb.nsheets)# 获取并打印 sheet 名称print( "sheet 名称:", wb.sheet_names())# 根据 sheet 索引获取内容sh1 = wb.sheet_by_index(0)# 或者# 也可根据 sheet 名称获取内容# sh = wb.sheet_by_name('成绩')# 获取并打印该 sheet 行数和列数print( u"sheet %s 共 %d 行 %d 列" % (sh1.name, sh1.nrows, sh1.ncols))# 获取并打印某个单元格的值print( "第一行第二列的值为:", sh1.cell_value(0, 1))# 获取整行或整列的值rows = sh1.row_values(0) # 获取第一行内容cols = sh1.col_values(1) # 获取第二列内容# 打印获取的行列值print( "第一行的值为:", rows)print( "第二列的值为:", cols)# 获取单元格内容的数据类型print( "第二行第一列的值类型为:", sh1.cell(1, 0).ctype)# 遍历所有表单内容for sh in wb.sheets():for r in range(sh.nrows):# 输出指定行print( sh.row(r))
我已经把每行代码都加了注释,应该可以很容易理解,接下来执行命令 python excel_r.py ,输出如下结果:
$ python excel_r.pysheet 数量: 2sheet 名称: ['成绩', '汇总']sheet 成绩 共 3 行 2 列第一行第二列的值为: 成绩第一行的值为: ['姓名', '成绩']第二列的值为: ['成绩', 88.0, 99.5]第二行第一列的值为: 1[text:'姓名', text:'成绩'][text:'张三', number:88.0][text:'李四', number:99.5][text:'总分'][number:187.5]
细心的朋友可能注意到,这里我们可以获取到单元格的类型,上面我们读取类型时获取的是数字1,那1表示什么类型,又都有什么类型呢?别急下面我们通过一个表格展示下:
| 数值 | 类型 | 说明 |
|---|---|---|
| 0 | empty | 空 |
| 1 | string | 字符串 |
| 2 | number | 数字 |
| 3 | date | 日期 |
| 4 | boolean | 布尔值 |
| 5 | error | 错误 |
通过上面表格,我们可以知道刚获取单元格类型返回的数字1对应的就是字符串类型。
修改 excel
上面说了写入和读取 Excel 内容,接下来我们就说下更新修改 Excel 该如何操作,修改时就需要用到 xlutils 中的方法了。直接上代码,来看下最简单的修改操作:
# excel_u.py# 导入相应模块import xlrdfrom xlutils.copy import copy# 打开 excel 文件readbook = xlrd.open_workbook("test_w.xls")# 复制一份wb = copy(readbook)# 选取第一个表单sh1 = wb.get_sheet(0)# 在第四行新增写入数据sh1.write(3, 0, '王亮')sh1.write(3, 1, 59)# 选取第二个表单sh1 = wb.get_sheet(1)# 替换总成绩数据sh1.write(1, 0, 246.5)# 保存wb.save('test_w1.xls')
从上面代码可以看出,这里的修改 Excel 是通过 xlutils 库的 copy 方法将原来的 Excel 整个复制一份,然后再做修改操作,最后再保存。现在我们执行以下命令 python excel_u.py 看下修改结果如下:
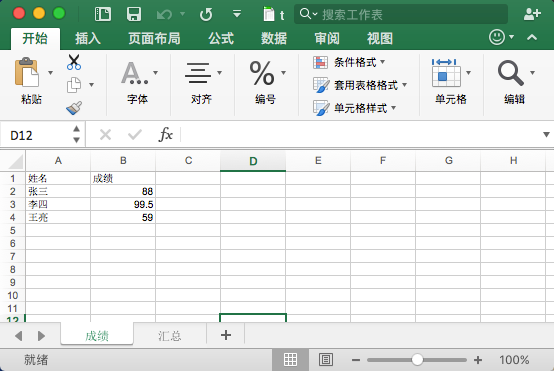

格式转换操作
在平时我们使用 Excel 时会对数据进行一下格式化,或者样式设置,在这里把上面介绍写入的代码简单修改下,使输出的格式稍微改变一下,代码如下:
# excel_w2.py# 导入 xlwt 库import xlwt# 设置写出格式字体红色加粗styleBR = xlwt.easyxf('font: name Times New Roman, color-index red, bold on')# 设置数字型格式为小数点后保留两位styleNum = xlwt.easyxf(num_format_str='#,##0.00')# 设置日期型格式显示为YYYY-MM-DDstyleDate = xlwt.easyxf(num_format_str='YYYY-MM-DD')# 创建 xls 文件对象wb = xlwt.Workbook()# 新增两个表单页sh1 = wb.add_sheet('成绩')sh2 = wb.add_sheet('汇总')# 然后按照位置来添加数据,第一个参数是行,第二个参数是列sh1.write(0, 0, '姓名', styleBR) # 设置表头字体为红色加粗sh1.write(0, 1, '日期', styleBR) # 设置表头字体为红色加粗sh1.write(0, 2, '成绩', styleBR) # 设置表头字体为红色加粗# 插入数据sh1.write(1, 0, '张三',)sh1.write(1, 1, '2019-01-01', styleDate)sh1.write(1, 2, 88, styleNum)sh1.write(2, 0, '李四')sh1.write(2, 1, '2019-02-02')sh1.write(2, 2, 99.5, styleNum)# 设置单元格内容居中的格式alignment = xlwt.Alignment()alignment.horz = xlwt.Alignment.HORZ_CENTERstyle = xlwt.XFStyle()style.alignment = alignment# 合并A4,B4单元格,并将内容设置为居中sh1.write_merge(3, 3, 0, 1, '总分', style)# 通过公式,计算C2+C3单元格的和sh1.write(3, 2, xlwt.Formula("C2+C3"))# 对 sheet2 写入数据sh2.write(0, 0, '总分', styleBR)sh2.write(1, 0, 187.5)# 最后保存文件即可wb.save('test_w3.xls')
然后我们执行命令 python excel_w2.py 运行以上代码,来输出文件 test_w3.xls ,我们来看看效果怎么样。
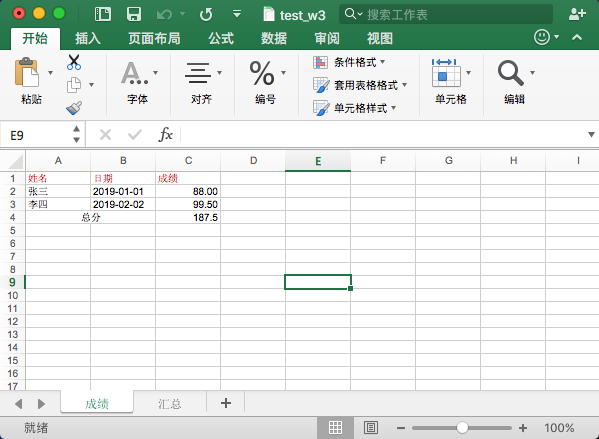
可以看出,使用代码我们可以对字体,颜色、对齐、合并等平时 Excel 的操作进行设置,也可以格式化日期和数字类型的数据。当然了这里我们只是介绍了部分功能,不过这已经足够我们日常使用了,想了解更多功能操作可以参考文末官网。
总结
本文为大家介绍了 Python 中如何操作 Excel 的常用类库,并详细介绍了下 xlrd & xlwt & xlutils 系列工具的使用,总体来看操作并不复杂,不过它对 xlsx 支持比较差,对修改其实支持也不太好,而且功能并不多,不过在 xls 操作中还是占有重要地位的,之后会为大家介绍其他常用 Excel 操作工具。