隆基泰和 做网站什么网站可以做医疗设备的
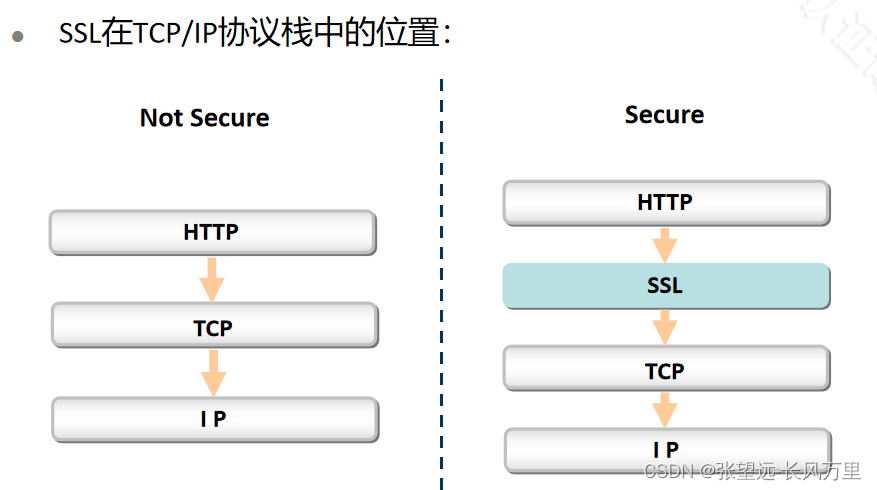
SSL的位置-思维导图
参考来源:
华为培训ppt:HCSCE122_SSL VPN技术
##SSL的位置
SSL协议套件
握手阶段,完成验证,协商出密码套件,进而生成对称密钥,用于后续的加密通信。
加密通信阶段,数据由对称加密算法来加解密。
密钥交换算法:保证对称密钥的交换是安全的,典型算法包括 DHE、ECDHE。
身份验证和签名算法:确认服务端的身份,其实就是对证书的验证,非对称算法就用在这里。典型算法包括 RSA、ECDSA。
补充:如果是双向验证(mTLS),服务端会验证客户端的证书。
对称加密算法:对应用层数据进行加密,典型算法包括 AES、DES。
消息完整性校验算法:确保消息不被篡改,典型算法包括 SHA1、SHA256。
典型SSL密钥套件的例子:
TLS_ECDHE_RSA_WITH_AES_128_CBC_SHA(0xc013)
- TLS 不用多说,代表了 TLS 协议。
ECDHE 是密钥交换算法,双方通过它就不用直接传输对称密钥,而只需通过交换双方生成的随机数等信息,就可以各自计算出对称密钥。
RSA 是身份验证和签名算法,主要是客户端来验证服务端证书的有效性,确保服务端是本尊,非假冒。
AES128_CBC 是对称加密算法,应用层的数据就是用这个算法来加解密的。这里的 CBC属于块式加密模式,另外一类模式是流式加密。
SHA 就是最后的完整性校验算法(哈希算法)了,它用来保证密文不被篡改。 0xc013
呢,是这个密码套件的编号,每种密码套件都有独立的编号。完整的编号列表在 IANA 的网站上可以找到。
过程全貌-思维导图

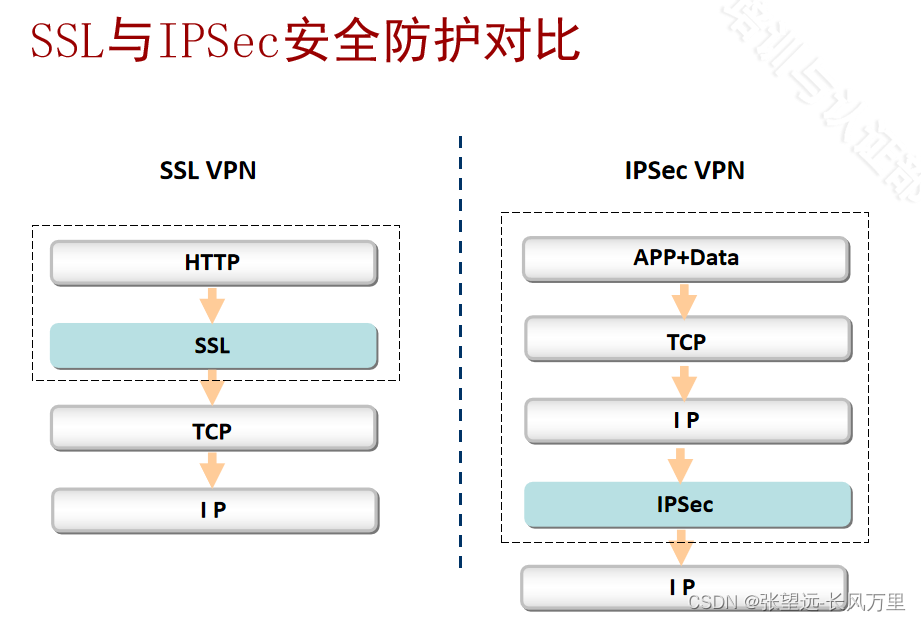
##对比ipsec